罕见病又称孤儿病,患病人数相对较少。不同地区对罕见病的定义有所不同,在我国,罕见病是指患病率在人群中低于1/500000或在新生儿中低于1/10000的疾病。
据估计,我国约有1680万罕见病患者,为帮助临床医生更好地认识这些罕见疾病,本文汇总了肝病科的5种罕见疾病,以飨各位同道。
一、肝豆状核变性
肝豆状核变性又称Wilson病(WD),是一种常染色体隐性遗传病。由ATP7B基因突变导致体内铜离子转运及排泄障碍,铜在肝脏、神经系统、角膜、肾脏等脏器蓄积,出现一系列临床表现。
WD主要发生在儿童和青少年中,可累及全身多个脏器。儿童患者多以肝脏受累为首发表现,青少年及成人以神经系统受累为首发症状的患者较多。肝脏病变可表现为无症状的转氨酶持续升高、慢性肝炎、肝硬化和急性肝功能衰竭。神经系统受累可表现为运动功能障碍、震颤、共济失调等,部分患者合并精神行为异常。其他受累系统包括肾脏、血液系统、骨关节、心脏、内分泌和生殖系统等。

图1 WD诊疗流程
二、遗传性血色病
遗传性血色病(HH)是一种常染色体隐性遗传病,男性多见,由先天性铁过多沉积于肝细胞、胰腺上皮细胞、心肌细胞、关节软组织细胞内等处引起相应脏器损伤的疾病。
HH首先累及肝脏,在MRI主要表现为“黑肝征”。然后累及胰腺,然后心肌,最后累及关节,出现心、肝、胰腺、甲状腺、性腺、皮肤关节等多器官并发症,包括皮肤色素沉着、肝肿大和肝硬化、糖尿病、性欲减退和睾丸萎缩、甲状腺功能减退、关节病变等。
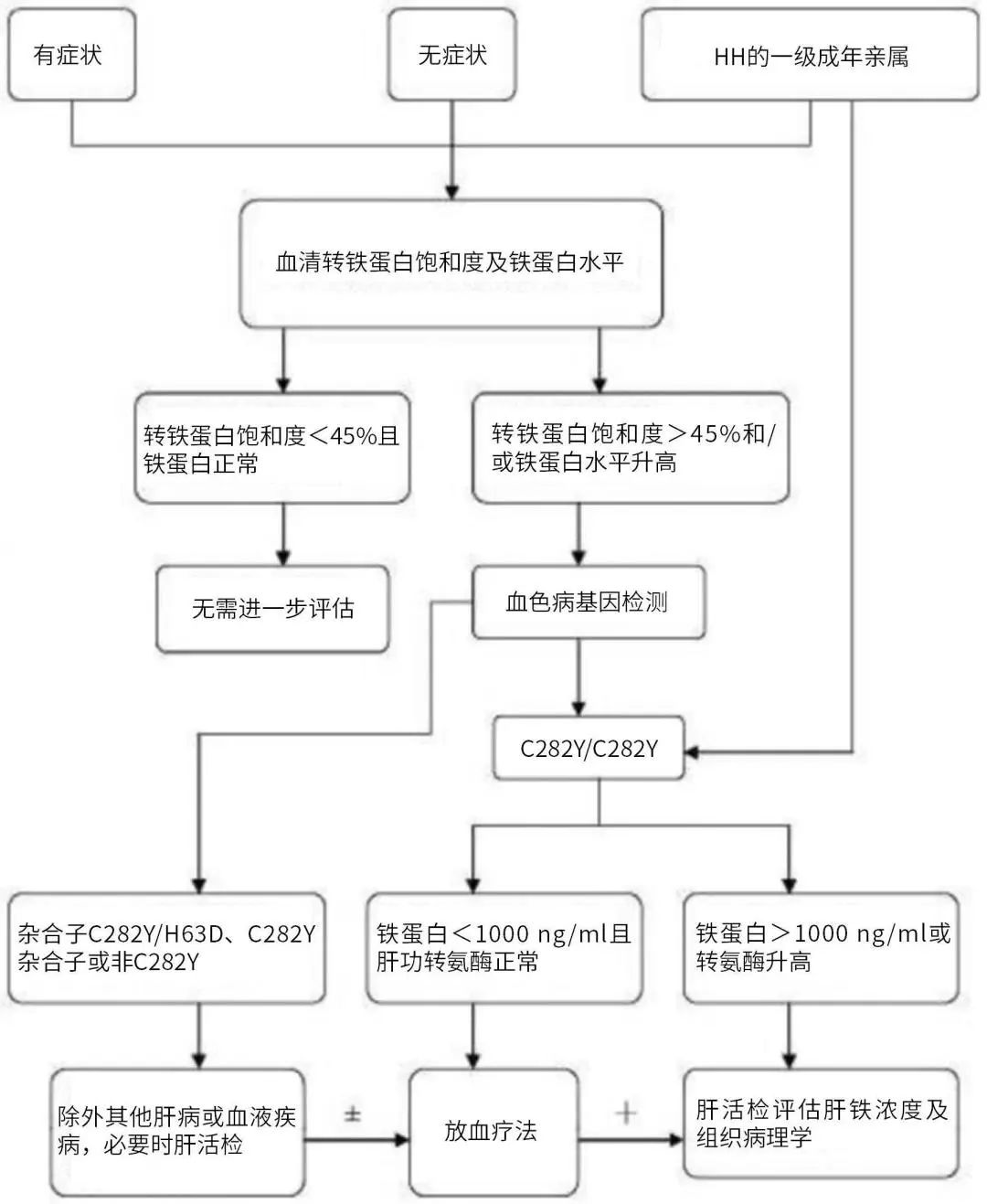
图2 HH诊疗流程
三、进行性家族性肝内胆汁淤积症
进行性家族性肝内胆汁淤积症(PFIC)是一组常染色体隐性遗传性疾病。因基因突变导致胆汁排泌障碍,发生肝内胆汁淤积,最终可发展为肝衰竭。根据致病基因不同,PFIC主要分为3型,包括PFIC-1型、PFIC-2型和PFIC-3型。
PFIC典型临床表现为黄疸和皮肤瘙痒。其他症状包括身材矮小、青春期发育落后等发育迟缓表现,胆囊结石,脂肪吸收障碍所致的脂肪泻,肝脾肿大,以及脂溶性维生素缺乏所致佝偻病、骨龄延迟、干眼症、凝血障碍和神经肌肉病变等症状。
PFIC的诊断依靠临床表现、血生化(血结合胆红素、碱性磷酸酶及胆酸等不同程度增高,血胆固醇多正常,PFIC-3型有血谷氨酰转肽酶增高)、胆汁成分分析、肝组织病理学检查以及基因检测等综合判断,并需要排除其他原因所致的胆汁淤积性肝病。
PFIC的治疗包括对症治疗(补充维生素、中链甘油三酯等,改善营养状态)、药物治疗[熊去氧胆酸10~30 mg/(kg·d)]、外科手术治疗(胆汁分流术)和肝移植。
四、先天性胆汁酸合成障碍
先天性胆汁酸合成障碍(IEBAS)是由于合成两种主要胆汁酸所必需的酶存在遗传缺陷,而导致的先天性胆汁酸合成障碍。IEBAS是一类常染色体隐形遗传病。
IEBAS的临床表现为进行性胆汁淤积性肝病、神经系统病变及脂溶性维生素吸收不良等。诊断需综合临床症状、实验室检查(结合胆红素升高、转氨酶升高、γ谷氨酸转肽酶正常)、辅助检查和病理活检,确诊依赖尿胆汁酸检测和基因检测。
对于IEBAS的治疗,如果诊断时已发生严重的肝功能损害,常需接受肝移植治疗;如果无严重的肝功能损害,则遵循个体化治疗原则,补充初级胆汁酸,根据尿中异常代谢产物含量判断疗效并调整治疗。
五、纯合子家族性高胆固醇血症
家族性高胆固醇血症(FH)是由低密度脂蛋白胆固醇(LDL-C)分解代谢的关键基因之一发生突变所引起的一种遗传性疾病。纯合子家族性高胆固醇血症(HoFH)是由于这些关键基因发生纯合突变或者复合性杂合突变所致。
HoFH的主要临床表现包括出生后即发现LDL-C水平明显升高,胆固醇沉积在皮肤、眼睛以及肌腱形成黄色瘤和脂性角膜弓。
HoFH的诊断需依据基因标准和临床标准。
基因诊断标准:通过基因检测发现两个等位基因存在LDLR、APOB、PCSK9或者LDLRAP1基因位点的突变。
临床诊断标准:在未治疗的情况下,LDL-C>500 mg/dl(>13 mmol/L)或者治疗后LDL-C>300 mg/dl(>8 mmol/L)以及以下情况之一∶10岁之前出现皮肤或者肌腱黄色瘤;父母LDL-C水平升高,符合杂合子FH的标准。
HoFH的治疗包括健康生活方式(推荐低饱和脂肪、低胆固醇、对心脏健康的饮食)、药物(主要为他汀类药物,可与其他降低胆固醇药物联合使用)、脂蛋白血浆清除、肝移植和其他手术治疗。